What has stood out the most when attempting to engage in the design of new proteins is that practically nothing can be seen.

All steps in the synthesising process, involve pipetting microlitres of what appears to be clear liquid from one vial to another, occassionally punctuated by tiny pellets of material which appear after the liquids are centrifuged.

Given the aim of my research project is to focus on the aesthetic generation of protein shape, it is ironic not to be able to see what we are attempting to create. All steps that I have described in the last two blogs, have first involved drawing diagrams on whiteboards or scraps of paper of intricate processes, such as DNA transcription and translation, before doing the actual step in the experimental process: ligating the plasmid, inserting the DNA fragment, or transforming bacterial cells for example.

At times synthesising proteins feels like an act of experimental faith: a blind adhesion to a new kind of alchemy that promises to transform liquid into life.

At a glance, it could be argued that designing synthetic proteins involves a radical cleaving of material processes from the experimental techniques of their making; that the language and graphics through which protein design is articulated is separate from the chemical-biological act of their physical manipulation. However, after each step in the synthesising process, a physical test is conducted to see if the procedure was successful. For example, to pipette the “products” potentially produced at various steps of the synthesising process into the wells for our gel tests, we added a coloured dye to be sure that we were able to see the micro-amount of liquid we were inserting.

And for the results to be seen in our tests, we added ethidium bromide to the gel which enters the DNA structure, allowing it to be seen under UV light. While these techniques rely on adding the means by which to visualise the micro-volume of reaction to the experimental product or to the material in which it will be tested, experimentally they at least indicate the material connection between their practical implementation and scientific articulation. Though at times the interpretation of the results is ambigous, these test functions in a similar way to those of forensics: evidence of events that have taken place are read from the physical marks which they leave.

However, no visual machinations of DNA manipulation are present in the actual laboratory process. Nothing of the intricate machinations of genetic manipulation and cellular transformations are be seen. No direct visualisations of the biochemical processes taking place are available in the way that might be understood for tissue culture for example, which uses a microscope to see material transformations of cells, and often video capture to record their changes.

When the experimental process is predominantly evidenced by trace, it begs the question: where does this leave protein design generally, and in particular relation to my project, its aesthetic and creative engagement?

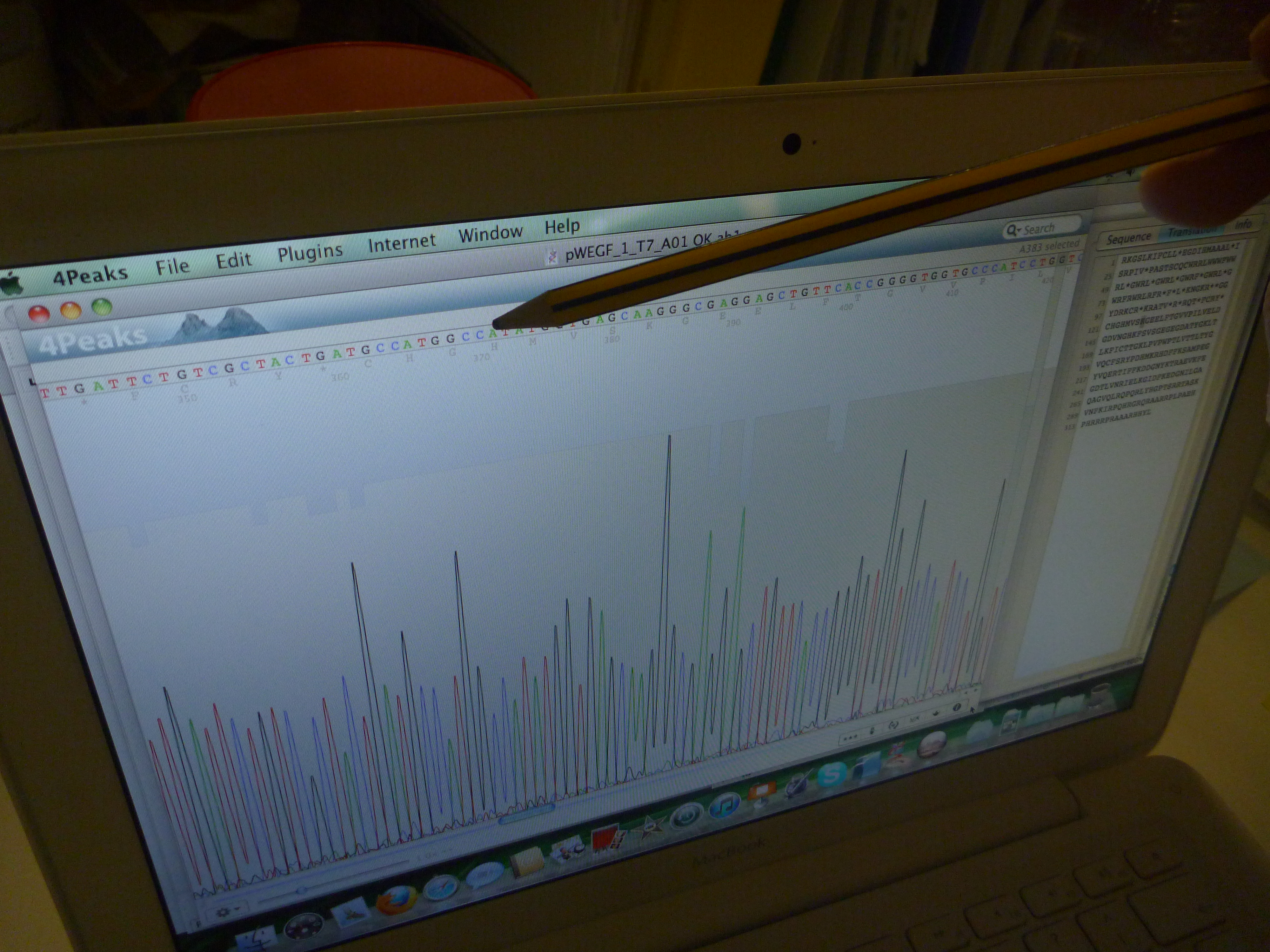
When we speak of design, it is commonly understood as a process which is ultimately expressed in a tangible way that translates what is developed on the computer, whiteboard, or drafting pad in to a physical functioning object: a building, a car, a piece of furniture. However, in protein design this process frequently occurs in silico, using scriptural and visual forms: the DNA code of GATC, and its corresponding expression as the 20 letters that code for amino acids, the visualisations of how stands these codes fold into the structural functional forms of proteins. But none of these processes can actually be seen in the minute amounts of material being manipulated during experiments. Well not often anyway. Xray crystallography can be used to visual the protein structure. Indeed, it is used to verify whether what has been designed has actually occured and matches with what was intended. However, it is not a readily usable research tool as it is extremely time-consuming and expensive, and so is effectively a procedure in itself that takes place after an experiment is conducted. On the basis, what is visualised through xray crystallography arguably doesn’t directly form part of the visualisation of protein syntheis, but is a trace of an experimental process.

How then, can we speak of design when design process takes place through textual and visual forms that are not part of regular laboratory practice, and cannot be evidenced through direct visualisation? What is the relation of the language and instruments of design when their actual implementation is invisible?







{kind=link}
{kind=link}
{kind=link}
{kind=link}